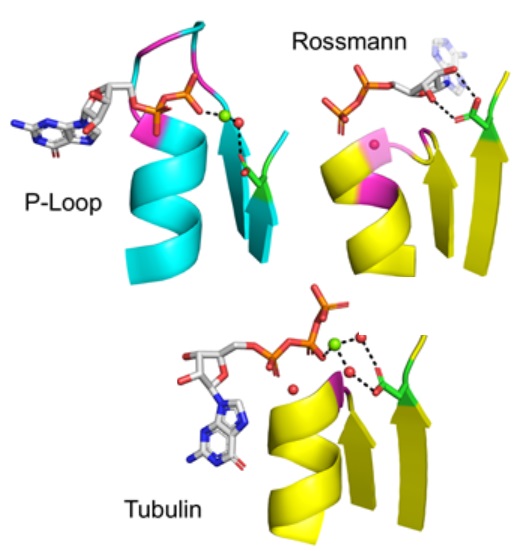
On
the emergence of P-Loop NTPase and Rossmann
enzymes from a Beta-Alpha-Beta ancestral
fragment
with Liam M Longo, Jagoda Jabłońska, Pratik
Vyas, Manil Kanade, Nir Ben-Tal, and Dan S
Tawfik, eLife (2020) pdf
Potential
Antigenetic Cross-reactivity Between Severe
Acute Respiratory Syndrome Coronavirus 2
(SARS-CoV-2) and Dengue Viruses
with Yaniv Lustig, Shlomit Keler, Nir Ben-Tal,
Danit Atias-Varon, Ekaterina Shlush, Motti
Gerlic, Ariel Munitz, Ram Doolman, Keren
Asraf, Liran I Shlush, and Asaf Vivante,
Clinical Infectious Diseases (2020) pdf
Potential
In Silico Structural and Biochemical
Functional Analysis of a Novel CYP21A2
Pathogenic Variant
with Michal Cohen, Emanuele Pignatti, Monica
Dines, Adi Mory, Nina Ekhilevitch, Christa E.
Flück, and Dov Tiosano, International Journal
of Molecular Sciences (2020) pdf
(our
rejected cover art)
On
the evolution of protein-adenine binding with Aya Narunsky, Amit Kessel,
Ron Solan, Vikram Alva, and Nir Ben-Tal PNAS(2020) pdf
To understand how
protein-ligand interactions emerged in
evolution, we analyzed all protein-adenine
complexes of known structure. All of
adenine's hydrogen donors and acceptors
may facilitate molecular recognition in
various binding modes, indicative of
convergent evolution. Furthermore,
adenine often binds to 'themes', segments
of amino acids that are commonly found in
proteins, and reported earlier.
Evolutionary
pathways of repeat protein topology in
bacterial outer membrane proteins with Meghan Franklin, Sergey
Nepomnyachyi, Ryan Feehan, Nir Ben-Tal, and
Joanna Slusky, eLife (2018) pdf
Image from Vikas Nanda's review
highlighting this work.
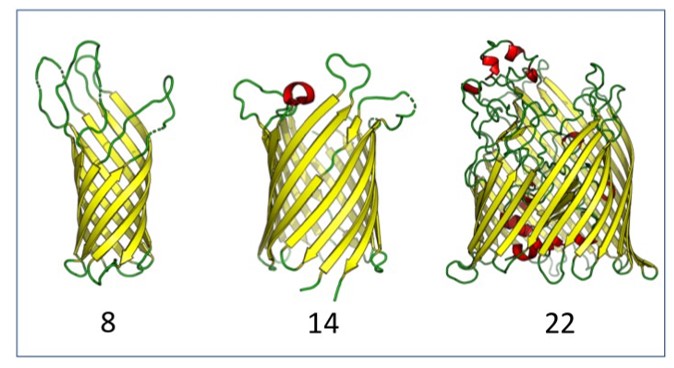
Outer
membrane proteins (OMPs) are the proteins in
the surface of Gram-negative bacteria. These
proteins have diverse functions but a single
topology: the β-barrel. Sequence analysis
has suggested that this common fold is a
β-hairpin repeat protein, and that
amplification of the β-hairpin has resulted
in 8–26-stranded barrels. Using an
integrated approach that combines sequence
and structural analyses, we find events in
which non-amplification diversification also
increases barrel strand number. Our
network-based analysis reveals
strand-number-based evolutionary pathways,
including one that progresses from a
primordial 8-stranded barrel to 16-strands
and further, to 18-strands. We also
find that the evolutionary trace is
particularly prominent in the C-terminal
half of OMPs, implicating this region in the
nucleation of OMP folding.
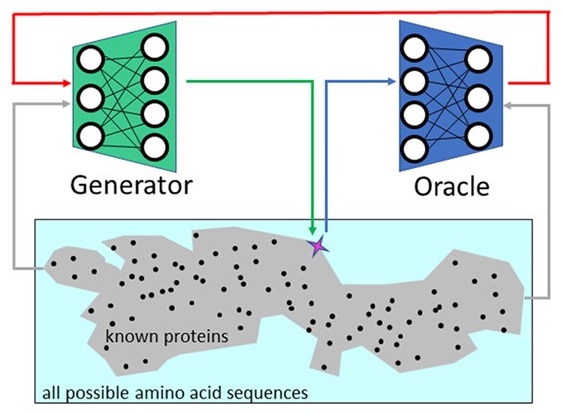
Navigating
Among Known Structures in Protein Space
with
Aya Narunsky and Nir Ben-Tal, Computational methods in
protein evolution (2019) pdf
Efflux
Pumps Represent Possible Evolutionary
Convergence onto the β-Barrel Fold
with
Meghan Whitney-Franklin, Sergey Nepomnyachiy,
Ryan Feehan, Nir Ben-Tal, and Joanna
S.G.Slusky, Structure
(2018) pdf
There are around
100 varieties of outer membrane
proteins in each Gram-negative
bacteria, all with the same
up-down β-barrel fold. Here we
suggest that like lysins, β-barrels of efflux
pumps have converged on this fold. By grouping
structurally solved outer membrane β-barrels
(OMBBs) by sequence we find that the membrane
environment may have led to convergent evolution
of the barrel fold. Specifically, the lack of
sequence linkage to other barrels coupled with
distinctive structural differences, such as
differences in strand tilt and barrel radius,
suggest that the outer membrane factor of efflux
pumps evolutionarily converged on the barrel.
Rather than being related to other OMBBs,
sequence and structural similarity in the
periplasmic region of the outer membrane factor
of efflux pumps suggests an evolutionary link to
the periplasmic subunit of the same pump
complex.
A
novel geometry-based approach to infer protein
interface similarity
with
Inbal Budowski-Tal and Yael Mandel-Gutfreund, Scientific Reports (2018)
pdf We present PatchBag – a geometry
based method for efficient comparison of
protein surfaces and interfaces. PatchBag is a
Bag-Of-Words approach, which represents
complex objects as vectors, enabling to search
interface similarity efficiently.
Complex
Evolutionary Footprints Revealed in an
Analysis of Reused Protein Segments of Diverse
Lengths
with
Sergey Nepomnyachiy and Nir Ben-Tal, PNAS
(2017) pdf
We question a central paradigm: namely, that
the protein domain is the "atomic unit" of
evolution. In conflict with the current
textbook view, our results unequivocally show
that duplication of protein segments happen
both above and below the domain level among
amino acid segments of diverse lengths.
Indeed, we show that significant evolutionary
information is lost when the protein is
approached as a string of domains. Our
finer-grained approach reveals a far more
complicated picture, where reused segments
often intertwine and overlap with each
other. Our results are consistent with a
recursive model of evolution, in which
segments of various lengths, typically smaller
than domains, "hop" between
environments. The fit segments remain,
leaving traces that can still be
detected.
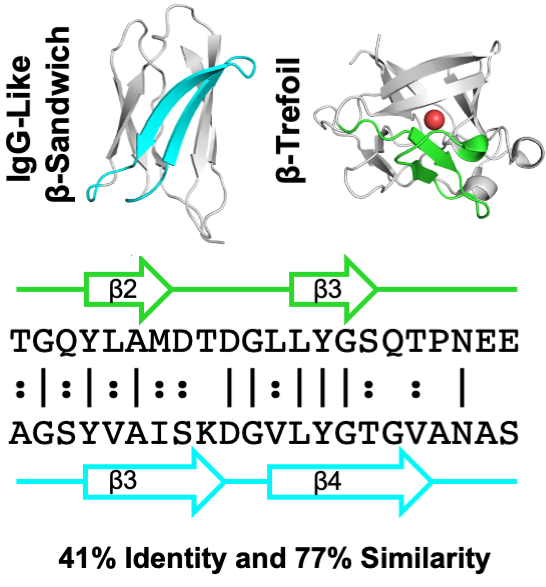
Similarity
between the Usher Plug and the Repeating
Domain of an Ice-adhesin:
Evolution via Surface Reshaping
with
Amit Kessel and Nir Ben-Tal, Israel
Journal of Chemistry (2017) pdf
The
PapC usher and MpAFP ice-adhesin feature
Ig-like domains, which are similar in shape
and sequence but are engaged in vry different
functions. We explore how evolution
reshaped the surfaces of these two domains to
fit to their respective functions.
ConTemplate
Suggests Possible Alternative Conformations
for a Query Protein of Known Structure
with
Aya Narunsky, Sergey Nepomnyachiy, Haim
Ashkenazy, and Nir Ben-Tal, Structure
(2015) pdf
Protein function involves conformational
changes, but often, for a given protein, only
some of these conformations are known. The
missing conformations could be predicted using
the wealth of data in the PDB. Most PDB
proteins have multiple structures, and
proteins sharing one similar conformation
often share others as well. The ConTemplate
web server
http://bental.tau.ac.il/contemplate)
exploits these observations to suggest
conformations for a query protein with at
least one known conformation (or model
thereof). We demonstrate ConTemplate on a
ribose-binding protein that undergoes
significant conformational changes upon
substrate binding. Querying ConTemplate with
the ligand-free (or bound) structure of the
protein produces the ligand-bound (or free)
conformation with a root-mean-square deviation
of 1.7A (or 2.2A); the models are derived from
conformations of other sugar-binding proteins,
sharing approximately 30% sequence identity
with the query. The calculation also suggests
intermediate conformations and a pathway
between the bound and free conformations.
CyToStruct:
Augmenting the Network Visualization of
Cytoscape with the Power of Molecular
Viewers
with
Sergey Nepomnyachiy, and Nir Ben-Tal, Structure
(2015) pdf
It can be informative to view biological data,
e.g., protein-protein interactions within a
large complex, in a network representation
coupled with three-dimensional structural
visualizations of individual molecular
entities. CyToStruct, introduced here,
provides a transparent interface between the
Cytoscape platform for network analysis and
molecular viewers, including PyMOL, UCSF
Chimera, VMD, and Jmol. CyToStruct launches
and passes scripts to molecular viewers from
the edges and nodes of the network. We provide
demonstrations to analyze interactions among
subunits in large protein/RNA/DNA complexes,
and similarities among proteins. CyToStruct
enriches the network tools of Cytoscape by
adding a layer of structural analysis,
offering all capabilities implemented in
molecular viewers. CyToStruct is available at
https://
bitbucket.org/sergeyn/cytostruct/wiki/Home
and in the Cytoscape App Store. Given the
coordinates of a molecular complex, our web
server (
http://
trachel-srv.cs.haifa.ac.il/rachel/ppi/ )
automatically generates all files needed to
visualize the complex as a Cytoscape network
with CyToStruct bridging to PyMOL, UCSF
Chimera, VMD, and Jmol.
Representation
of the protein universe using
classifications, maps, and networks
with
Nir Ben-Tal Israel
Journal of Chemistry (2014) pdf A meaningful
and coherent global picture of the protein
universe is needed to better understand protein
evolution and the underlying biophysics. We
survey the studies that tackled this fundamental
challenge, providing a glimpse of the protein
space. A global picture represents all known
local relationships among proteins, and needs to
do so in a comprehensive and accurate manner.
Three types of global representations can be
used: classifications, maps, and networks. In
these, the local relationships are derived,
based on the similarity of the proteins’
sequences, structures, or functions (or a
combination of these). Alternatively, the local
relationships can be co-occurrences of elements
in the protein universe. The representations can
be based on different objects: full polypeptide
chains, fragments, such as structural domains,
or even smaller motifs. Different protein
qualities were revealed in each study; many
point out the uniqueness of domains of the
alpha/beta SCOP (structural classification of
proteins) class.
Published in special issue to celebrate Michael
Levitt's Nobel prize.
Global
view of protein evolution
with
Sergey Nepomnyachiy, and Nir Ben-Tal Proc.
Natl. Acad. Sci. (USA) (2014)
pdf
We've been mentioned in PNAS
Highlights (text from there)
Just as the elements in the periodic table can
be traced back to the Big Bang, the set of all
proteins in terrestrial organisms reflects the
history of evolution on Earth. A global view
of this so-called protein universe would help
reveal how proteins evolve and are related to
one another, but empirical evidence exists for
relatively few relationships between proteins.
Sergey Nepomnyachiy et al. applied network
theory to a representative set of all known
protein domains drawn from the Structural
Classification of Proteins (SCOP) database.
The authors represented protein space using
two network configurations: a domain network
in which edges connect domains the segments of
which share similar sequence and structural
motifs, and a motif network in which edges
connect recurring motifs that lie within the
same domains. The authors demonstrate how
networks suggest evolutionary paths between
domains and provide clues about the mechanisms
of protein evolution. The findings offer an
approach to representing protein space that
could aid protein design, according to the
authors.
Redundancy-weighting
for better inference of protein structural
features
with
Chen Yanover, Natalia Vanetik, Michael Levitt,
and Chen Keasar Bioinformatics
(2014) pdf
In this study we explore the concept of
redundancy-weighted data-sets, originally
suggested by Miyazawa and Jernigan.
Redundancy-weighted data-sets include all
available structures and associate them (or
features thereof) with weights that are
inversely proportional to the number of their
homologs. Here, we provide the first
systematic comparison of redundancy-weighted
data-sets with non-redundant ones. We test
three weighting schemes and show that the
distributions of structural features that they
produce are smoother (having higher entropy)
compared with the distributions inferred from
non-redundant data-sets. We further show that
these smoothed distributions are both more
robust and more correct than their
non-redundant counterparts.We suggest that the
better distributions, inferred using
redundancy-weighting, may improve the accuracy
of knowledge-based potentials, and increase
the power of protein structure prediction
methods. Consequently, they may enhance
model-driven molecular biology.Â
On
the Universe of Protein Folds
with
Leonid Pereyaslavets,
Abraham O. Samson, and Michael Levitt Ann.
Rev. of Biophysics (2013) pdf
In the fifty years since the first atomic
structure of a protein was revealed, tens of
thousands of additional structures have been
solved. Like all objects in biology, proteins
structures show common patterns that seem to
define family relationships. Classification of
proteins structures, which started in the
1970s with about a dozen structures, has
continued with increasing enthusiasm, leading
to two main fold classifications, SCOP and
CATH, as well as many additional databases.
Classification is complicated by deciding what
constitutes a domain, the fundamental unit of
structure. Also difficult is deciding when two
given structures are similar. Like all of
biology, fold classification is beset by
exceptions to all rules. Thus, the
perspectives of protein fold space that the
fold classifications offer differ from each
other. In spite of these ambiguities, fold
classifications are useful for prediction of
structure and function. Studying the
characteristics of fold space can shed light
on protein evolution and the physical laws
that govern protein behavior.
From
Protein Structure to Function via
Computational Tools and Approaches
with
Mickey Kosloff Isr.
J. Chem. (2013)pdf
The 3D structures of proteins are often
considered fundamental for understanding their
function. Yet, because of the complexity of
protein structure, extracting specific
functional information from structures can be
a considerable challenge. Here, we present
selected approaches and tools that were
developed in the Kolodny and Kosloff labs to study
and connect protein sequence, structure, and function spaces.Â
Maps
of protein structure space reveal a
fundumental relationship between protein
structure and function
with
Margarita Osadchy Proc.
Natl. Acad. Sci. (2011)
108(30):12301-6 pdf
We've been mentioned in f1000
We propose a new method to efficiently create
three-dimensional maps of structure space
using a very large data set of > 30,000
SCOP domains.Â
In our maps, each domain is represented
by a point, and the distance between any two
points approximates the structural distance
between their corresponding domains. We
use these maps to study the spatial
distributions of properties of proteins, and
in particular those of local vicinities in
structure space such as structural density and
functional diversity. These
maps provide a novel broad view of protein
space, and thus reveal new fundamental
properties thereof. At
the same time, the maps are consistent with
previous knowledge (e.g., domains cluster by
their SCOP class), and organize in a unified,
coherent representation previous observation
concerning specific protein folds. To
investigate the function-structure
relationship, we measure the functional
diversity (using the Gene Ontology controlled
vocabulary) in local structural vicinities. Our
most striking finding is that functional
diversity varies considerably across structure
space: the space has a highly diverse region,
and diversity abates when moving away from it. Interestingly,
the domains in this region are mostly
alpha/beta structures, which are known to be
the most ancient proteins.Â
A
library of protein surface patches
discriminates between native structures and
decoys generated by structure prediction
servers
withRoiGamliel, KlaraKedem, and Chen Keasar BMC
Structural Biology (2011) 11:20.online version
FragBag, a
"bag-of-words" representation of protein
structure, retrieves structural neighbors
from the entire PDB quickly and accurately
with
InbalBudowski-Tal and Yuval
Nov Proc.
Natl. Acad. Sci.(2010)
107: 3481-3486 pdf ,web-page
In FragBag, we
describe a protein structure by the collection
of its overlapping short contiguous backbone
segments, and discretize
this set using a library of fragments (using
our libraries described below).
Then, we represent the protein as a
‘bags-of-fragments’ – a vector that
counts the number of occurrences of each
fragment – and measure the similarity
between two structures by the similarity
between their vectors.
We use ROC curve analysis to
quantify the success of FragBag
in identifying neighbor candidate sets in a
dataset of over 2,900 structures. The gold
standard is the set of neighbors found by six
state-of-the-art structural aligners (the same
data set from our comparison study). Our
best FragBag
library finds more accurate candidate sets
than three other filter methods: SGM, PRIDE,
and a method by Zotenkoet
al. More interestingly, FragBag performs on a
par with the computationally expensive, yet
highly trusted, structural aligners STRUCTAL
and CE .
Sequence-Similar,
Structure-dissimilar protein pairs in the
PDB
with
Mickey Kosloff Proteins:
Structure, Function, and Bioinformatics (2007)
71(2):
891-902 pdf, database
It is often assumed that in the Protein Data
Bank (PDB), two proteins with similar
sequences will also have similar structures.
This assumption underlies many computational
studies and structure prediction methods.
Here, we compare sequence-based structural superpositions and
geometry-based structural alignments and show
that the former provides a better measure of
structure dissimilarity. Using sequence-based
structural superpositioning
we find many examples in the PDB where two
proteins that are similar in sequence have
structures that differ significantly from one
another, usually in direct relation to their
function. We conclude that the assumption of
two proteins with similar sequences having
similar structures is often incorrect and can
lead to the loss of structurally and
functionally important information.
VISTAL
- A two-dimensional visualization tool for
structural alignments
with
Barry Honig Bioinformatics
(2006) 22(17):
2166-2167 pdf, software
VISTAL describes structures as a series of
secondary structure elements, and places
matched residues one on top of each other
colored according to the three-dimensional
distance of their Ca atoms.
Using
an Alignment of Fragment Strings for
Comparing Protein Structures.
with
Iddo Friedberg,
Tim Harder, EinatSitbon, Zhanwen Li, and Adam Godzik Bioinformatics
(2006) 23(2):
e219-e224 pdf
This
work by Iddo and
Tim, compares
protein structures that are described via
strings of fragments from our libraries.
Protein
Structure Comparison: Implications for the
Nature of 'Fold Space', and Structure and
Function Prediction.
with Donald Petrey and Barry Honig Curr. Opin. Struct. Bio.
(2006) 16:
393-398 pdf
We argue in favor of viewing protein structure
space as continuous, with potential structural
similarities between any pair of structures.
This is different from the traditional
perspective in which a structure is in a
particular group (denoted fold) and only other
structures within that fold are considered as
its structural neighbors. We survey recent
progress made in the prediction of protein
structure and function by relying on these
relationships.
Faster
Algorithms for Optimal Multiple Sequence
Alignment based on Pairwise
Comparisons.
withPankaj K. Agarwal and Yonatan Bilu
Lecture Notes in Computer Science (WABI 2005)
3692: 315-327 2005.
pdf,
online
material
We consider the following version of the
Multiple Sequence Alignment (MSA) problem: In
a preprocessing stage pairwise
alignments are found for every pair of
sequences. The goal is to find an optimal
alignment in which matches are restricted to
positions that were matched at the
preprocessing stage. We present several
techniques for making the dynamic programming
algorithm more efficient, while still finding
an optimal
solution under these restrictions. In our
formulation the MSA must conform with pairwise (local)
alignments, and in return can be solved more
efficiently. We prove that it suffices to find
an optimal alignment of sequence segments,
rather than single letters, thereby reducing
the input size and thus improving the running
time.
Comprehensive
Evaluation of Protein Structure Alignment:
Scoring by Geometric Measures.
with Patrice
Koehl and Michael
Levitt J.
Mol. Biol. (2005) 346,
1173-1188. pdf,
online
material
We report a comprehensive comparison of
protein structural alignment methods.
Specifically, we evaluate six publicly
available structure alignment programs: SSAP,
STRUCTAL, DALI, LSQMAN, CE and SSM by aligning
all 8,581,970 protein structure pairs in a
test set of 2,930 sequence diverse protein
domains. We follow the traditional path and
rely on a gold standard (the CATH
classification) and compare the rates of true
and false positives using ROC curves. However,
due to limitations of this methodology, we
also compare the alignments directly, using
geometric match measures.
Inverse
Kinematics in Biology: The Protein Loop
Closure Problem.
withLeonidasGuibas, Michael Levitt
and Patrice Koehl Int.
Jour. Robotics Research. (2005)
24,
151-162. pdf
We address an inverse kinematics problem in
structural biology: the loop closure problem.
We describe a procedure for generating the
conformations of candidate loops that fit in a
gap in a protein structure framework. Our
method concatenates small fragments of protein
from small libraries of representative
fragments. Our approach has the advantages of
ab initio
methods since we are able to enumerate all
candidate loops in the discrete approximation
of the conformational space accessible to the
loop, as well as the advantages of database
search approach since the use of fragments of
known protein structures guarantees that the
backbone conformations are physically
reasonable.
Approximate
Protein Structural Alignment in Polynomial
Time.
with Nathan Linial Proc.
Natl. Acad. Sci., (2004) 101 (33),
12201-12206. pdf,
online
material
Protein structural alignment is a fundamental
problem in computational structural biology.
Here, we study it as a family of optimization
problem and provide a polynomial time
algorithm to solve them. We also show an
NP-hardness proof of an alternative approach
to this problem using internal distance
matrices. Lastly, we visualize the scoring
function for several pairs of structures.
Protein
Decoy Assembly Using Short Fragments Under
Geometric Constraints.
with Michael
Levitt Biopolymers,
(2003) 68,
278-285. pdf
We use the libraries of fragments described
below to generate decoys for several proteins.
Coupled with a descriminating
energy function, decoys are useful for
predicting protein structure. It seems that
this method works well for all alpha proteins.
Small
Libraries of Protein Fragments Model Native
Protein Structures Accurately.
with Patrice
Koehl, LeonidasGuibas and Michael
Levitt J.
Mol. Biol. (2002) 323,
297-307. pdf,
online
material
We study efficient means of modeling protein
structure. Our model concatenates elements
from libraries of commonly observed protein
backbone fragments into approximate
structures. There are no additional degrees of
freedom so a string of fragment labels fully
defines a three-dimensional structure; the set
of all strings defines the set of structures
(of a given length). By varying the size of
the library and the length of its fragments,
we generate structure sets of different
resolution. With larger libraries, the
approximations are better, but we get good
fits to real proteins (less than 1A) with less
than 5 states per residue.